In computational chemistry, we often want to estimate the amount of energy some physical process will take. Example processes are ligand binding to a protein receptor, transferring a small molecule from gas to water, or mutating some side chain in a protein. The quantity we are looking for is called the free energy, i.e., the energy in a system that is free to do work. (This is opposed to, for example, a system’s overall internal energy or its “unusable” energy measured by entropy.) Free energies are relative values, not absolute values. We can compare energies of some altered state relative to a reference state, but individual values by themselves don’t mean anything. This is why values are often reported as the free energy difference corresponding to some physical process.*
Obtaining free energy differences is not a simple subtraction of state A free energy minus state B free energy.** We can get these from a number of different approaches [1], broadly categorized:
- Probability densities
- Free energy perturbation
- Thermodynamic integration
- Nonequilibrium work
Going back to the main goal, we want to find the free energy change from state A to state B. The thing about running computer experiments, is that we’re not constrained to modeling real-life physical processes. Free energy is a state variable, meaning that it doesn’t matter how state A gets to state B – it’s path independent. The reason we might not want to model real-life processes is that it’s computationally infeasible. A typical timestep used in a molecular dynamics simulation is 2 femtoseconds. That’s a quadrillionth of a second (10-15 s)! Running a simulation for several days might get you tens of nanoseconds. However, a typical protein-ligand binding event can take microseconds to milliseconds.[2] That would mean months-long simulations just to see that one event occur.† Even still, it’s hard to get a picture of the binding mechanism from a single long simulation. You’d need the simulation to be much longer than the time for association/dissociation so that you could see many binding/unbinding transitions happen. This is why we use techniques such as alchemical transformations or machine learning.
Footnotes
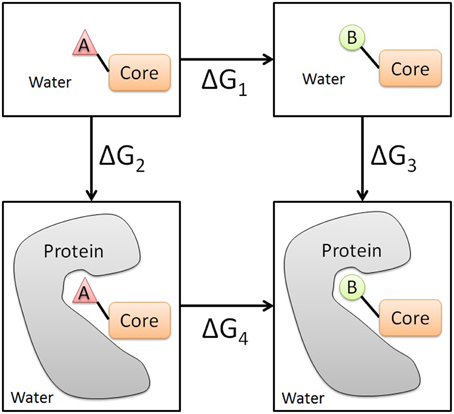
* You may see mention of absolute binding free energies, but these are still not absolute free energies in and of themselves. Thinking about a ligand binding to a protein, relative binding free energies refer to either transforming ligand A into ligand B inside the protein, or mutating some residue on the protein given the same ligand. See example thermodynamic cycles here and here, respectively. (Thermodynamic cycles seem alluringly simple, but don’t skimp on the mental power!) The term absolute in this context means that no transformation is taking place. The energies are still relative because we need to also consider ligand solvation and protein solvation. For example, we must consider the energy placing the ligand in solution instead of having it disappear completely. Here’s an illustrative thermodynamic cycle.
{kind=link}
{kind=link}
{kind=link}
** This goes into statistical mechanics some, but a simplified explanation is that the free energy of a system doesn’t just depend on the positions and momenta of the system (also known as its “phase space coordinates”). Rather, free energy depends on a quantity known as the partition function (PF). The partition function is to statistical mechanics what the wave function is to quantum mechanics. The PF is a sum of all accessible states that a system can be in; it represents a measure of the number of accessible states at some given temperature. From the PF, we can directly relate macroscopic properties (e.g., entropy, pressure) of the system to microscopic properties (e.g., particle masses, microstate energies)
† Computional power and resources are constantly getting better. With the relatively recent application of GPUs to computational sciences, this could mean decreased computational cost, although there’s still a ways to go.
Additional resources
- What is “free” energy?
- Calculation of binding free energies
- Time, the forgotten dimension of ligand binding teaching
References